Batch Processing
from nomspectra.spectrum import Spectrum
from nomspectra.spectra import SpectrumList
import nomspectra.draw as draw
import pandas as pd
import matplotlib.pyplot as plt
import os
Load spectra
We can load separate Spectrum, treat them and then join it in SpectrumList object which is a list of spectra
specs = SpectrumList()
for filename in sorted(os.listdir("data/similarity/")):
if filename[-3:] != 'csv':
continue
spec = Spectrum.read_csv(f"data/similarity/{filename}", assign_mark=True)
specs.append(spec)
specs.get_names()
['a_1', 'a_2', 'a_3', 'a_4', 'a_5', 'a_6']
Or directly load from folder if specs already treated
specs = SpectrumList.read_csv('data/similarity/')
specs.get_names()
['a_4', 'a_5', 'a_6', 'a_2', 'a_3', 'a_1']
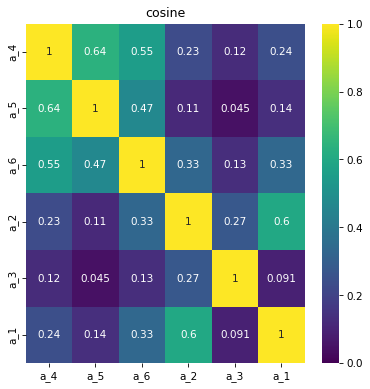
Calculate simmilarity index and plot matrix
Calculate simmilarity indexes. For now it common indexes - Cosine, Tanimoto and Jaccard
specs.get_simmilarity(mode='cosine')
array([[1. , 0.63921734, 0.55387418, 0.22893115, 0.12221844,
0.24206235],
[0.63921734, 1. , 0.46713676, 0.11426236, 0.04536192,
0.13553428],
[0.55387418, 0.46713676, 1. , 0.3297159 , 0.12996979,
0.33440804],
[0.22893115, 0.11426236, 0.3297159 , 1. , 0.27330141,
0.59910651],
[0.12221844, 0.04536192, 0.12996979, 0.27330141, 1. ,
0.0912144 ],
[0.24206235, 0.13553428, 0.33440804, 0.59910651, 0.0912144 ,
1. ]])
And plot matrix
specs.draw_simmilarity(mode='cosine')
Calculate metrics
From spectra we can get molecular metrics
specs.get_mol_metrics()
| a_4 | a_5 | a_6 | a_2 | a_3 | a_1 | |
|---|---|---|---|---|---|---|
| AI | -0.079344 | -0.037731 | -0.307631 | 0.444909 | 0.613860 | 0.171642 |
| C | 21.476279 | 21.143970 | 17.786572 | 22.186035 | 23.129087 | 17.967260 |
| CAI | 9.251700 | 8.712936 | 9.954005 | 15.300819 | 16.397214 | 12.064931 |
| CRAM | 0.552194 | 0.540266 | 0.485958 | 0.090364 | 0.035393 | 0.204550 |
| DBE | 12.644497 | 12.636119 | 8.106037 | 13.660797 | 16.722827 | 8.526630 |
| DBE-O | 0.806469 | 0.492241 | 0.560464 | 7.055005 | 10.396550 | 2.820194 |
| DBE-OC | 0.033105 | 0.017236 | 0.017788 | 0.298224 | 0.447654 | 0.118010 |
| DBE_AI | 0.419919 | 0.205085 | 0.273470 | 6.775581 | 9.990954 | 2.624301 |
| H | 20.004788 | 19.264393 | 21.578437 | 19.305284 | 15.211308 | 21.029147 |
| H/C | 0.941654 | 0.924041 | 1.220249 | 0.918817 | 0.655002 | 1.261374 |
| N | 0.341225 | 0.248690 | 0.217368 | 0.254807 | 0.398788 | 0.147887 |
| NOSC | 0.225485 | 0.268255 | -0.290940 | -0.289653 | -0.039698 | -0.601656 |
| O | 11.838028 | 12.143877 | 7.545574 | 6.605792 | 6.326276 | 5.706435 |
| O/C | 0.555214 | 0.575824 | 0.439298 | 0.295497 | 0.279618 | 0.315787 |
| S | 0.045326 | 0.038467 | 0.069625 | 0.024617 | 0.006808 | 0.048007 |
| Unnamed: 0 | 5134.428335 | 1918.021934 | 1496.312157 | 841.325051 | 1782.231948 | 1584.499424 |
| errorPPM | 0.001824 | 0.029912 | 0.045206 | -0.029267 | -0.030579 | 0.000557 |
| formula | NaN | NaN | NaN | NaN | NaN | NaN |
| mass | 473.590881 | 472.240469 | 361.207872 | 395.774294 | 399.951346 | 331.795644 |
| peakNo | 5134.428335 | 1918.021934 | 1496.312157 | 841.325051 | 1782.231948 | 1584.499424 |
| z | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 |
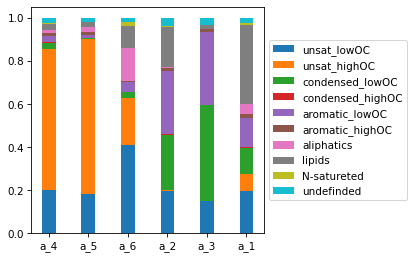
Get molecular class density and plot bar
specs.draw_mol_density()
specs.get_mol_density()
| a_4 | a_5 | a_6 | a_2 | a_3 | a_1 | |
|---|---|---|---|---|---|---|
| unsat_lowOC | 0.201447 | 0.183535 | 0.408356 | 0.195555 | 0.147491 | 0.196795 |
| unsat_highOC | 0.654447 | 0.716404 | 0.217642 | 0.006134 | 0.004349 | 0.080142 |
| condensed_lowOC | 0.026479 | 0.005946 | 0.028384 | 0.256306 | 0.441213 | 0.118184 |
| condensed_highOC | 0.002802 | 0.000989 | 0.000662 | 0.001651 | 0.002156 | 0.002919 |
| aromatic_lowOC | 0.028453 | 0.013982 | 0.044924 | 0.290728 | 0.336764 | 0.138210 |
| aromatic_highOC | 0.015343 | 0.011668 | 0.005026 | 0.014576 | 0.014677 | 0.016288 |
| aliphatics | 0.014729 | 0.023744 | 0.155616 | 0.005297 | 0.000097 | 0.045678 |
| lipids | 0.028270 | 0.022411 | 0.101287 | 0.186821 | 0.018383 | 0.369506 |
| N-satureted | 0.003950 | 0.000688 | 0.016474 | 0.005117 | 0.000000 | 0.006827 |
| undefinded | 0.024080 | 0.020633 | 0.021629 | 0.037814 | 0.034871 | 0.025451 |
Also we can calculate density of squares of Van Krevelen diagram
specs.get_square_vk()
| a_4 | a_5 | a_6 | a_2 | a_3 | a_1 | |
|---|---|---|---|---|---|---|
| 1 | 0.008942 | 0.004066 | 0.014149 | 0.046670 | 0.178541 | 0.009918 |
| 2 | 0.002034 | 0.001793 | 0.012627 | 0.033494 | 0.149713 | 0.005776 |
| 3 | 0.000819 | 0.000160 | 0.010486 | 0.046734 | 0.061893 | 0.010208 |
| 4 | 0.023089 | 0.017808 | 0.038597 | 0.041165 | 0.023237 | 0.069177 |
| 5 | 0.005045 | 0.003707 | 0.044065 | 0.153976 | 0.004767 | 0.293159 |
| 6 | 0.028251 | 0.009248 | 0.023760 | 0.356841 | 0.348926 | 0.188009 |
| 7 | 0.070833 | 0.049838 | 0.102810 | 0.199036 | 0.165541 | 0.093670 |
| 8 | 0.087435 | 0.083649 | 0.225485 | 0.060352 | 0.023909 | 0.103503 |
| 9 | 0.015644 | 0.021632 | 0.129569 | 0.008620 | 0.001865 | 0.045593 |
| 10 | 0.002791 | 0.006214 | 0.034856 | 0.006054 | 0.000000 | 0.027571 |
| 11 | 0.066727 | 0.092062 | 0.017016 | 0.035408 | 0.032921 | 0.039697 |
| 12 | 0.372026 | 0.420460 | 0.109793 | 0.010715 | 0.008141 | 0.040661 |
| 13 | 0.248249 | 0.219455 | 0.138720 | 0.000269 | 0.000090 | 0.048610 |
| 14 | 0.015856 | 0.013362 | 0.040365 | 0.000000 | 0.000000 | 0.011880 |
| 15 | 0.000423 | 0.001304 | 0.005874 | 0.000000 | 0.000000 | 0.001774 |
| 16 | 0.003531 | 0.008137 | 0.001184 | 0.000000 | 0.000211 | 0.002251 |
| 17 | 0.028628 | 0.036910 | 0.005151 | 0.000182 | 0.000000 | 0.003421 |
| 18 | 0.015810 | 0.010031 | 0.004591 | 0.000237 | 0.000123 | 0.003076 |
| 19 | 0.002573 | 0.000165 | 0.024572 | 0.000249 | 0.000000 | 0.001212 |
| 20 | 0.000423 | 0.000000 | 0.008698 | 0.000000 | 0.000000 | 0.000089 |
SpectrumList is a list
With SpectrumList object we can work as with list, for example, plot spectrum
for spec in specs:
draw.spectrum(spec)
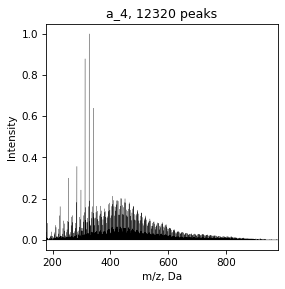
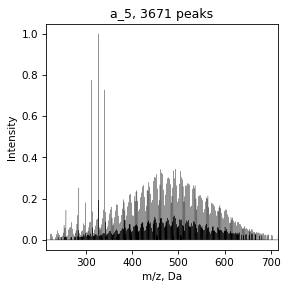
And save all data in folder
if 'temp' not in os.listdir():
os.mkdir('temp')
specs.to_csv('temp')